PPI3D Tutorial
This page shows the usage of PPI3D web server with an example of single-sequence search. The user interface of the two-sequences search mode is very similar and should be also clear after trying out the following example.
For more details on the interpretation of PPI3D results, please refer to the Help page.
The tutorial uses the sequence of DNA sliding clamp from E. coli, which can be obtained in FASTA format from UniProt![]() .
.
Input
Open the single-sequence search form from the navigation menu in the left:
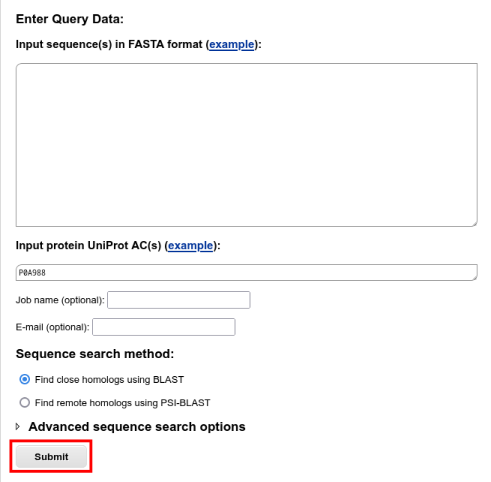
Input your query sequence.
You can paste the FASTA-formatted ![]() sequence into the input form, or put the UniProt AC of the query (P0A988) into the UniProt AC field.
To run a search using default BLAST options, press the "Submit" button:
sequence into the input form, or put the UniProt AC of the query (P0A988) into the UniProt AC field.
To run a search using default BLAST options, press the "Submit" button:
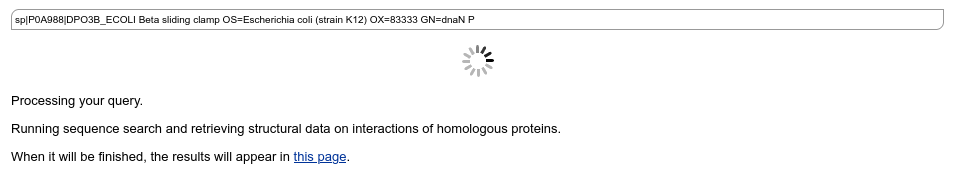
Query processing
BLAST search is fast and usually takes a very short time. While the job is running, PPI3D demonstrates a job progress page, which is automatically refreshed every few seconds:
Results summary
After the search finishes and the results are saved into the server's backend database, the results summary page appears.
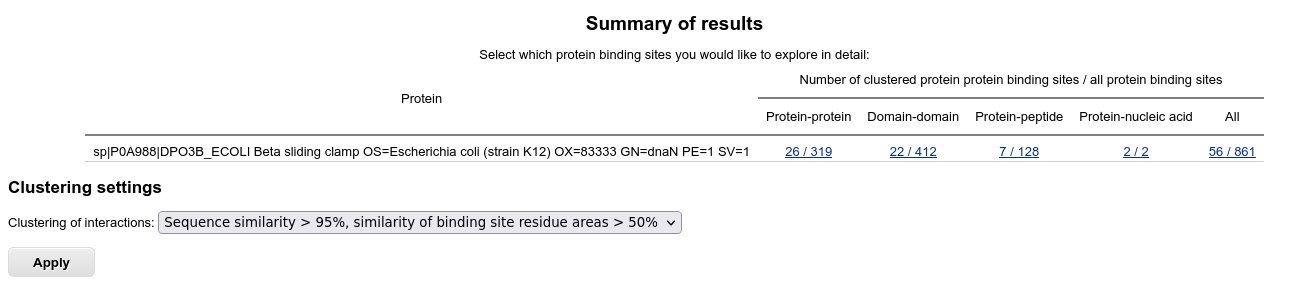
The table displays the summary of identified binding sites for the homologs of the query protein. To check the clustering options and control further output, you can select several clustering options. For example, choosing "Sequence similarity > 40%, similarity of binding site residue areas > 50%" allows grouping of similar interfaces together:

Analysis of clustered results
Analysis of homomeric interactions
To analyze homomeric protein-protein interactions, click on the numbers in "Protein-protein" column:

A table with the clustered results is opened. It can be filtered interactively by typing criteria into the fields under header row. Such rapid browser-side filtering allows to see only homomeric protein-protein binding sites, having large interfaces:
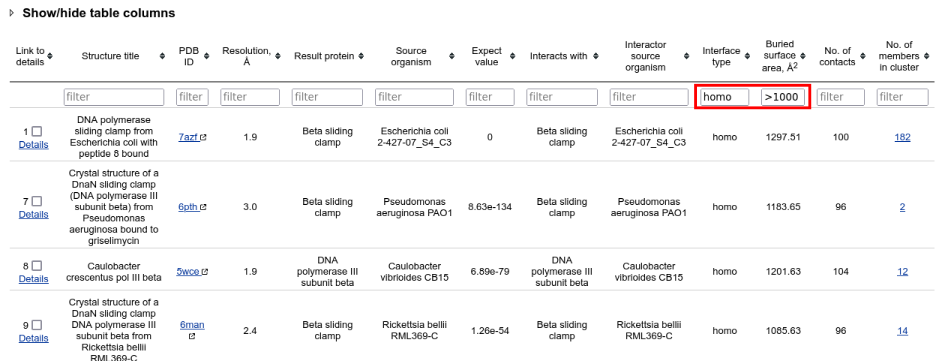
To summarize the binding sites from different clusters, select all visible rows and click "Summarize selected interactions" button below the table:

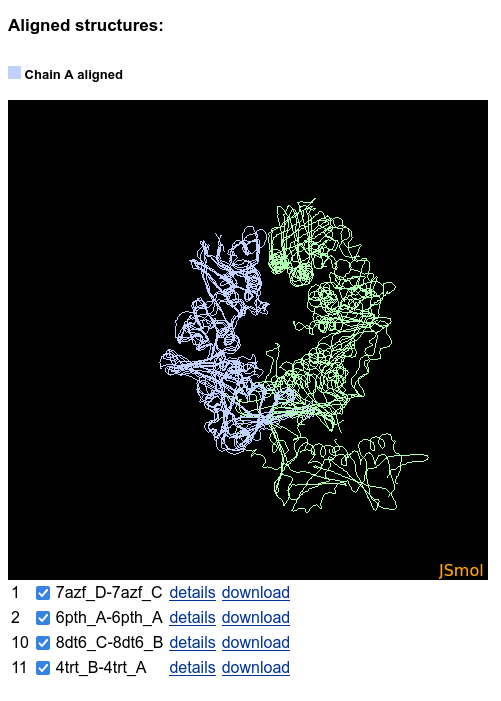
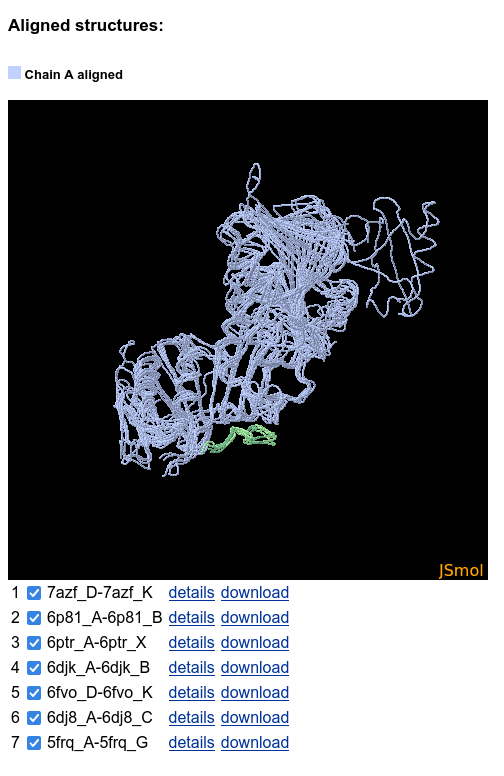
A page with selected sequences aligned to the query sequence is opened in new tab. The interface residues are marked and allow to identify which proteins having similar or different binding modes. Select some of the results to further align the corresponding structures:

A JSmol applet apears below the sequences table. It shows the aligned structures, and the alternative binding modes are clearly visible:
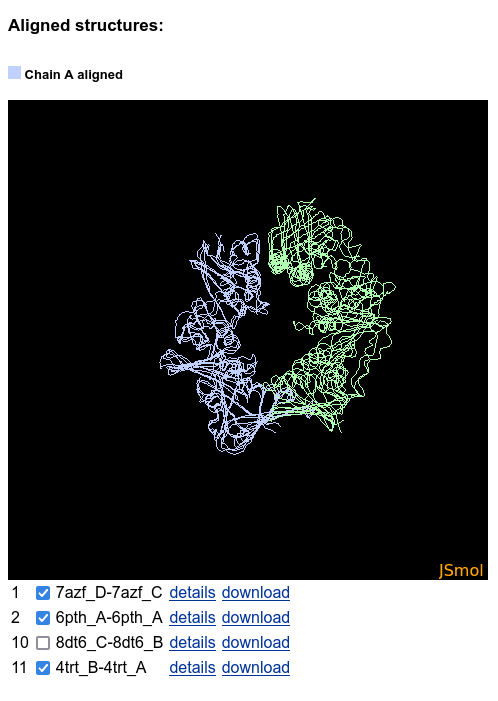
The structures can be hidden and shown using the checkboxes below the JSmol:
Analysis of heteromeric interactions
To analyze heteromeric interactions, return to the "Results summary" page:
Then choose protein-protein interactions again to see the clustered results table.
Heteromeric interactions of the DNA sliding clamps involve both proteins and peptides. Select both interaction types for display in the menu below the table and press "Apply":

Filtering the table to contain only heteromeric interactions and by other criteria is the same as in the example with homomeric interactions above. After a few easy steps you can summarize also heteromeric interfaces to see that all the peptides are bound to the same binding site:
Analysis of protein-nucleic acid interactions
Return to the "Results summary" page and select the protein-nucleic acid interactions from the table:

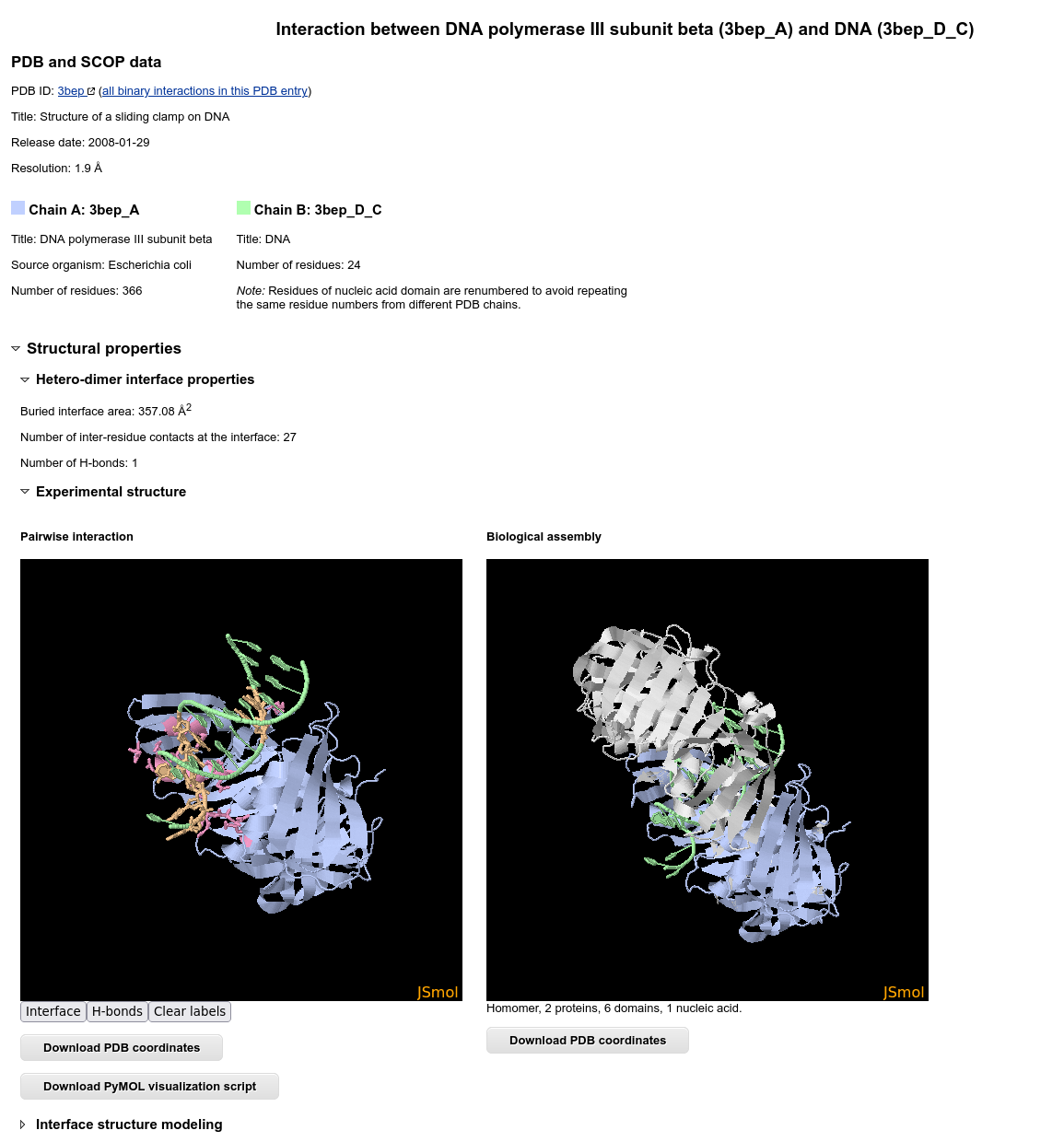
In the resulting table you can access the most detailed information on the interaction interface by clicking "Details" in the first column:

Analysis of the details of interaction interface
Structure analysis
On the top of the interaction details page, PDB entry and protein annotations displayed, followed by the structures in JSmol:
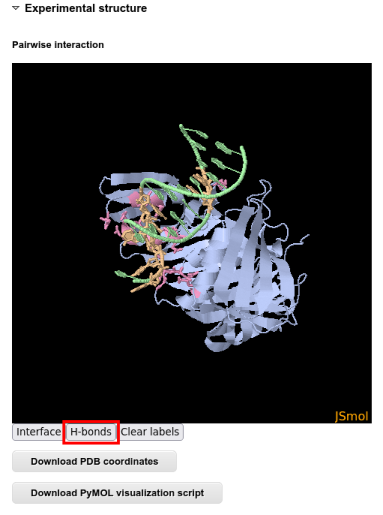
The structure visualization in JSmol is interactive and allows seeing different contact types:
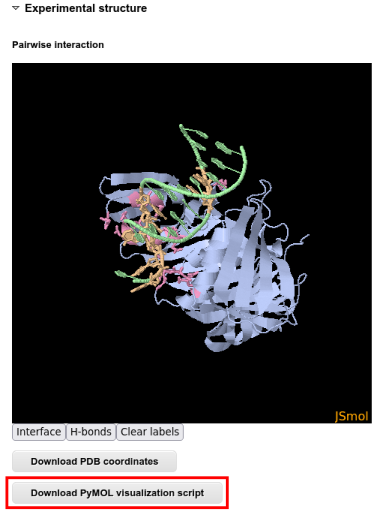
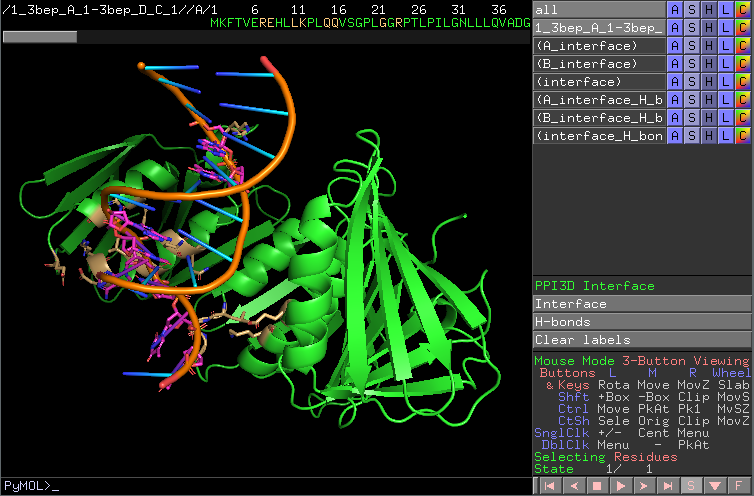
For a even more interactive analysis it is possible to download a PyMOL script and analyze the interaction interface in PyMOL:
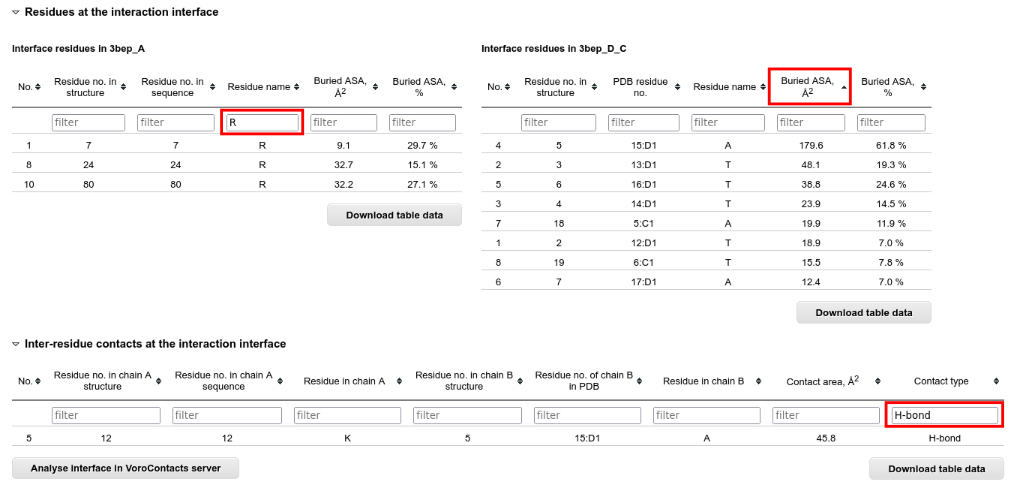
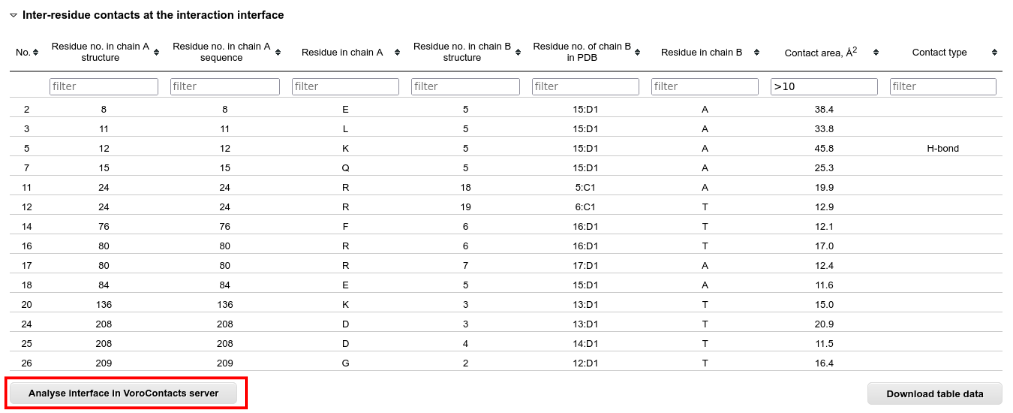
Below the JSmol applet there are the tables with interface residues and contacts. These tables can be sorted and filtered according to all columns:
For the analysis of the interface contacts at atom level, you can submit the structure to the VoroContacts ![]() server directly from PPI3D.
server directly from PPI3D.
Sequence alignment analysis
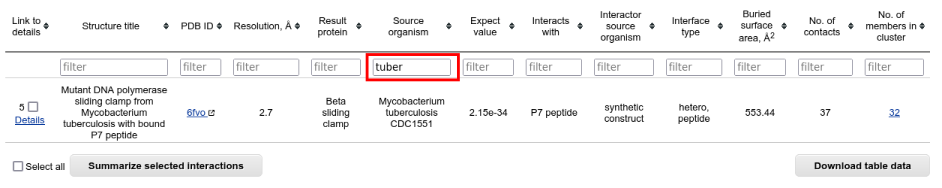
At the time of writing, structures of the protein-nucleic acid complexes are not available for the homologs of E. coli DNA sliding clamp, only for the query protein itself. Therefore to get more familiar with the analysis of sequence alignments you should get back to the "Results summary" page, select "Protein-peptide interactions" and choose the M. tuberculosis sliding clamps:
Then go the the "Interaction details" page, and see the sequence alignment in the bottom. The interface residues are colored and allow homology-based inference of the binding site. The alignment also shows the high conservation of the peptide binding site:
